“Vakar bija izsaukums uz ledus halli. Reanimējām 22 gadus vecu puisi. Viņš spēlēja hokeju, kārtējo spēli aizvadījis labi, ne par ko nebija sūdzējies, ne ar ko nav slimojis, bet tad dušā nokrita bezsamaņā – un viss.” “Dalot zāles, atrada, ka viena slimniece neelpo un ķermenis pavisam auksts. Palātas kaimiņienes neko nav manījušas, it kā bija mierīgi gulējusi pa nakti. Viņa bija ļoti jauka sieviete, tikai 58 gadus veca. Pirms trim gadiem bijis infarkts, ārstējās ar stenokardiju, dakteris jau gribēja rakstīt mājās.” Šādi gadījumi diemžēl sastopami klīniskajā praksē – un nebūt ne tik reti. Pašreizējas zināšanas ļauj saskatīt pēkšņas nāves paaugstināta riska pacientus. Pareizai riska novērtēšanai īpaši svarīgi ir atšķirt “snaudošas” bīstamas slimības pazīmes no līdzīgām labdabīgu slimību izpausmēm, ko dažkārt pat uzskata par normas variantu.
Pēkšņa kardiāla nāve (PKN) ir nāve sirds mehāniskas darbības pēkšņas apstāšanās dēļ, un tās pamatā ir sirds slimības. Visbiežāk šādu nāvi apraksta kā dabisku un pēkšņu, saistītu ar ātru samaņas zudumu, un tā iestājas nepilnu stundu pēc akūtu simptomu sākšanās. [1] Tiešs PKN iemesls visbiežāk ir kambaru ritma traucējumi - kambaru tahikardijas un/vai kambaru fibrilācijas. Taču ir jāsaprot, ka vairākumā gadījumu šādas nāves precīzu laiku un iemeslu ir grūti noteikt.
PKN iemesli dažādās vecumgrupās atšķiras:
- pacientiem pēc 35gadu vecuma PKN visbiežāk izraisa koronārā sirds slimība (KSS);
- pirms 35 gadu vecuma KSS arī ir iespējama, piemēram, iedzimtu koronāru asinsvadu anomāliju vai vielmaiņas traucējumu dēļ, kā arī lietojot dažādas toksiskas vielas. Taču gados jaunākiem pacientiem KSS ir novērojama daudz retāk. Viņiem PKN iemesls parasti ir citas sirds slimības, lielākoties pārmantotas.
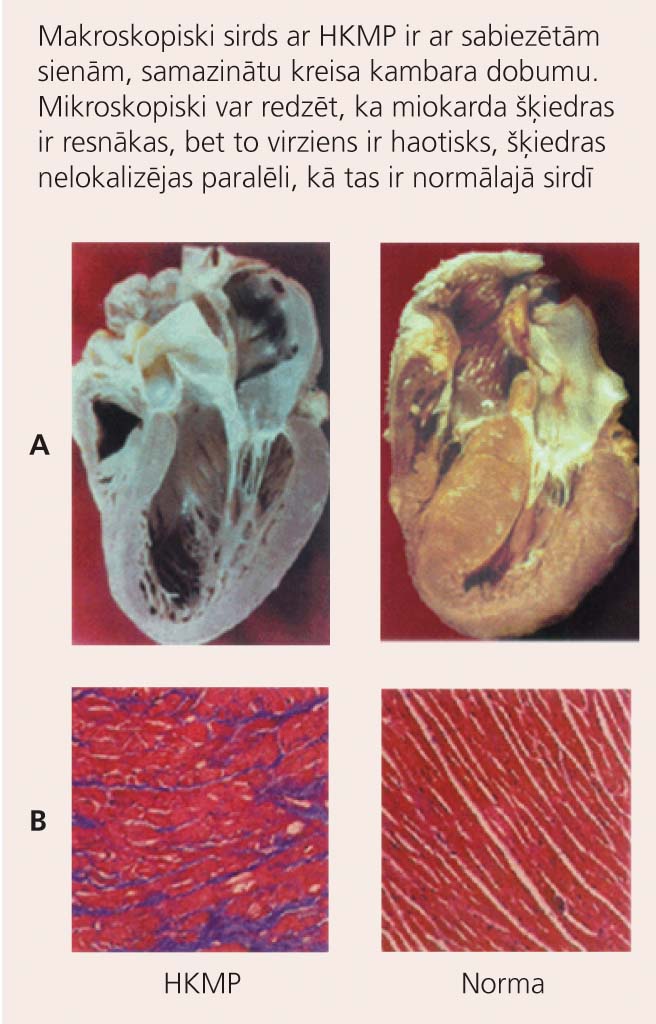
1. attēls
Makroskopisks (A) un mikroskopisks (B) sirds izskats pie HKMP (pa kreisi) un veselā sirdī (pa labi)
Iedzimtas sirds saslimšanas
Šā apskata tēma ir PKN pacientiem pirms 35 gadu vecuma jeb ar KSS nesaistīta PKN. Raksta mērķis ir atgādināt par dažām iedzimtām sirds saslimšanām, kas saistās ar paaugstinātu PKN risku, un izstāstīt par ieteicamo taktiku šādiem pacientiem, kā arī dalīties pieredzē par gadījumiem, kas nesen diagnosticēti Latvijā.
Sirds patoloģijas, kas var izraisīt PKN:
- hipertrofiska kardiomiopātija,
- dilatācijas kardiomiopātija,
- aritmogēna labā kambara displāzija,
- iedzimts gara QT intervāla sindroms,
- Brugada sindroms,
- kateholamīnu atkarīga polimorfa kambaru tahikardija.
Visas šīs slimības apvieno daži ļoti būtiski aspekti:
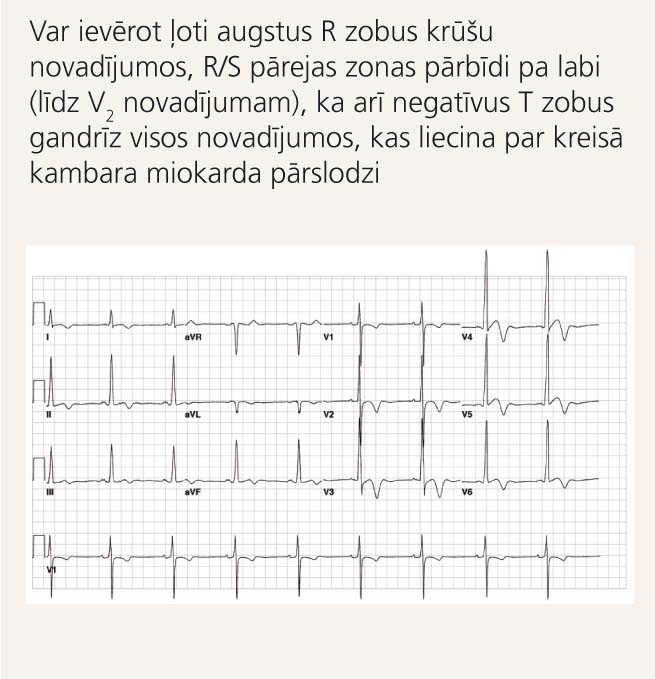
2. attēls
EKG pacientam ar HKMP
- tās ir ģenētiski noteiktas,
- to pirmā un diemžēl arī pēdējā izpausme var būt PKN,
- gandrīz visiem pacientiem ar šīm slimībām, veicot vienkāršus izmeklējumus, var konstatēt kādu no specifiskām pazīmēm, kas var norādīt uz patoloģijas esamību,
- ja patoloģisks process skar ļoti plašu sirds daļu un ir ar izteiktām klīniskām izpausmēm- hronisku sirds mazspēju vai sirds ritma traucējumiem-, vienīgais radikālais risinājums ir sirds transplantācija.
Šīs slimības vieno arī viens ieteikums no ārstēšanas un PKN profilakses viedokļa. Proti, pacientiem ar pierādītu hipertrofisku kardiomiopātiju, dilatācijas kardiomiopātiju, aritmogēnu labā kambara displāziju, iedzimtu garā QT intervāla sindromu, Brugada sindromu vai kateholamīnu atkarīgu polimorfu kambaru tahikardiju kategoriski aizliegts nodarboties ar sportu vai veikt smagus fiziskus darbus.
Līdzīgi jebkurām citām ģenētiski noteiktām slimībām arī šīm ir būtiski izmeklēt ne tikai pacientu, bet arī viņa radiniekus. Svarīgi ir izmeklēt PKN upurus un viņu ģimenes locekļus. Tādējādi var izvairīties no daudziem citiem nepatīkamiem gadījumiem.
Hipertrofiska kardiomiopātija (HKMP)
Šī ir visbiežākā ģenētiski noteiktā sirds saslimšana, un, spriežot pēc ehokardiogrāfisko pētījumu rezultātiem, tā ir 1 no 500 cilvēkiem vispārējā populācijā. [2]
Slimības pamatā ir defekts kādā no gēniem, kas kodē kardiomiocītu sarkomēra olbaltumvielas (miozīna smagās ķēdes proteīnu, aktīnu, tropomiozīnu u.c.). Rezultātā miokarda šķiedras palielinās un maina formu, tās ir dezorientētas (skat. 1. attēlu). Daļa no kardiomiocītiem iet bojā, un tos aizvieto fibroblasti un starpšūnu saistaudi. Šo izmaiņu sekas ir lokāli elektriska impulsa vadīšanas traucējumi, kas dod iespēju attīstīties kambaru tahikardijām vai pat fibrilācijām. [3]
Lielākajai daļai pacientu HKMP norit asimptomātiski. Simptomi, kas var būt saistīti ar šo slimību, ir aizdusa, diskomforts krūtīs, galvas reiboņi, sirdsklauves un ģīboņi. Bet dažreiz pirmais HKMP simptoms ir PKN. Jāpiemin, ka visbiežāk novērojama PKN saistībā ar fizisku slodzi.
Diagnostika
Slimības diagnostikā zelta standarts ir ehokardiogrāfiska izmeklēšana, kas būtu indicēta visiem pacientiem, kam ar jau pieminētajām HKMP klīniskajām pazīmēm ir arī izmaiņas 12 novadījumu elektrokardiogrammā (EKG), kas liecina par kreisā kambara hipertrofiju.
EKG par kreisā kambara hipertrofiju var liecināt (skat. 2. attēlu) [4]:
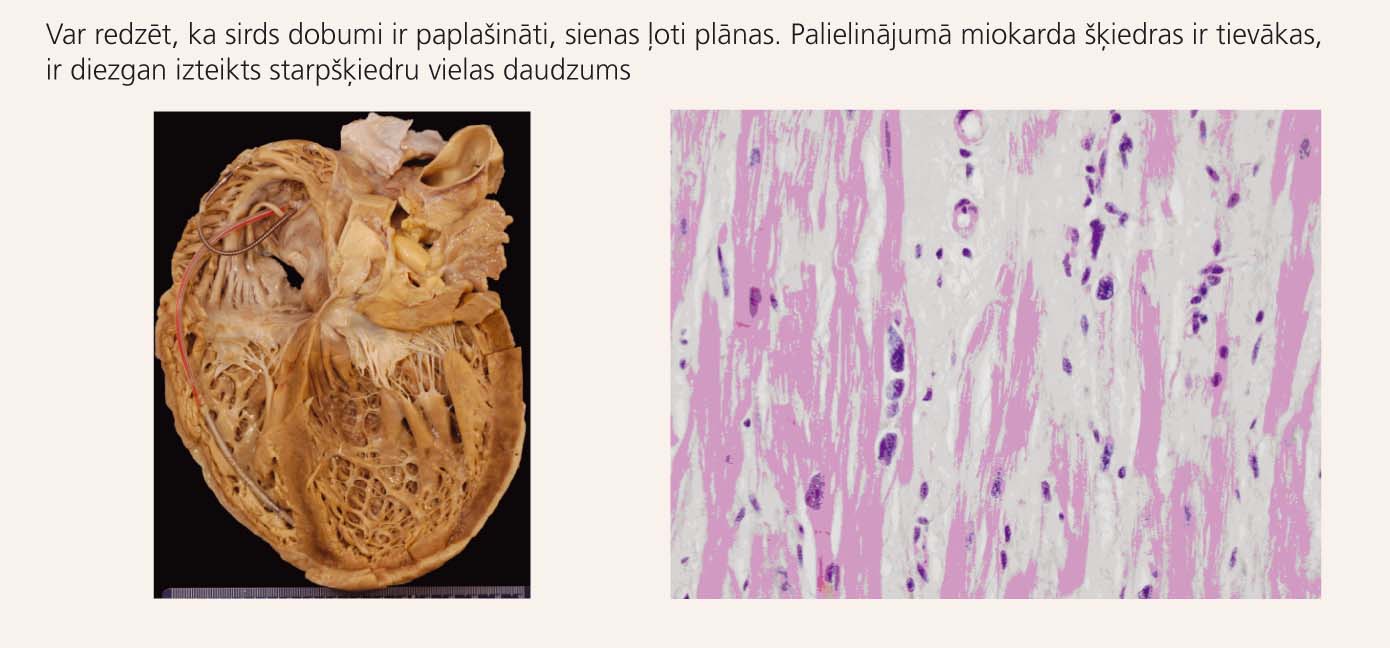
3. attēls
Makroskopisks (pa kreisi) un mikroskopisks (pa labi) sirds izskats pie DKMP
- R zoba amplitūdas V5vai V6novadījumos+ Szoba amplitūdas V1vai V2novadījumos summa >35mm (Sokolow-Lyon kritērijs);
- kopējā QRSvoltāža (Q+R+S zobu summa) visos 12novadījumos >180mm;
- S zoba amplitūdas V3novadījumā+ Rzoba amplitūdas aVLnovadījumā summa >28mm vīriešiem un >20mm sievietēm (Kornela kritērijs);
- R zoba amplitūda novadījumos V5vai V6>26mm;
- R zoba amplitūda novadījumā aVL>11mm;
- R zoba amplitūda novadījumā aVF>20mm;
- ST-T segmenta noslīdējums, kas liecina par miokarda pārslodzi.
Ir pacientu grupa, kam HKMP diagnostika ir īpaši sarežģīta, - tie ir sportisti un cilvēki, kas strādā smagu fizisku darbu. Slodzes ietekmē arī notiek miokarda hipertrofija, bet tā ir fizioloģiska un nav saistīta ar PKN risku. Dažkārt ir ļoti grūti izlemt, vai konstatētās izmaiņas šiem pacientiem ir patoloģijas pazīmes vai fizioloģiskas adaptācijas rezultāts.
Ārstēšana
Ieteikumi HKMP pacientu ārstēšanai: [6]
- asimptomātiskiem pacientiem un pacientu radiniekiem regulāri jāveic ehokardiogrāfija. Atbilstīgi hipertrofijas pakāpei, pacienta vecumamu.c. rādītājiem tā jāveic reizi 6-12vai pat 60mēnešos;
- ja pacientam progresē kreisā kambara hipertrofija vai sirds mazspējas klīniskās izpausmes, tālākai ārstēšanai pacients jāsūta pie speciālista;
- pacients ar HKMP neatliekami jāsūta pie speciālista, ja ir bijuši ģīboņi, īpaši fiziskas slodzes laikā;
- nepieciešamības gadījumā jāizšķiras par beta-adrenoblokatoru lietošanu;
- atsevišķos gadījumos tiek lietotas citas ārstēšanas metodes:
- kambaru starpsienas alkohola ablācija vai ķirurģiska septāla miektomija pie izolētas kambaru starpsienas hipertrofijas ar kreisā kambara izejas trakta obstrukciju,
- implantējama kardiovertera-defibrilatora (ICD) implantācija dzīvībai bīstamu kambaru aritmiju gadījumos,
- specifiska pastāvīga divkameru elektrokardiostimulatora implantācija.
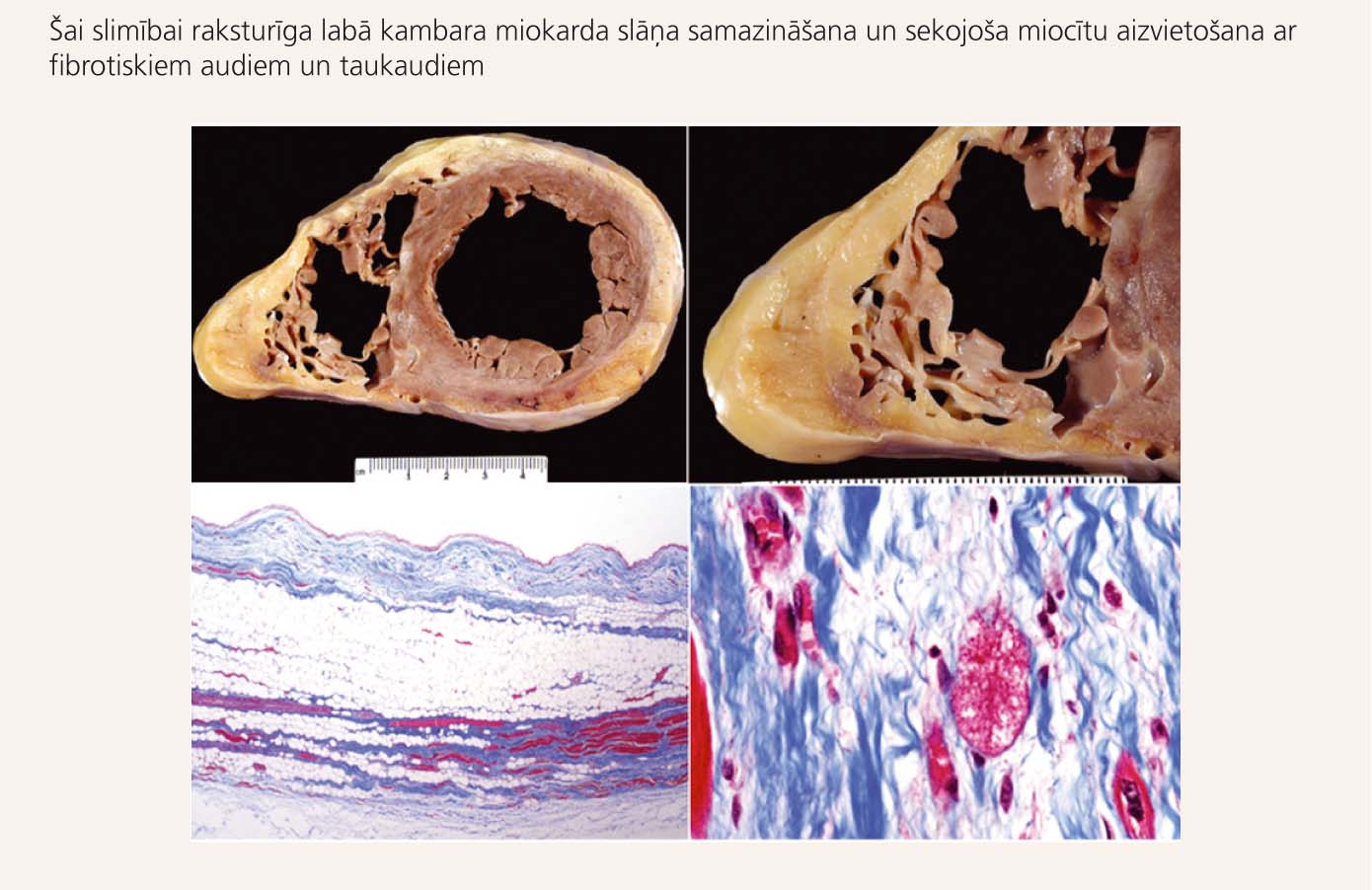
4. attēls
Makroskopisks (augšējie preparāti) un mikroskopisks (apakšējie preparāti) sirds izskats pie ALKD
Gadījums Latvijā
Sešpadsmit gadu vecs vīrietis, iepriekš ne ar ko nebija slimojis. Daudz nodarbojies ar sportu, pusprofesionālā līmenī spēlējis hokeju, regulāri nodarbojies ar skriešanu, īpaši ar hokeju saistītajās vasaras nometnēs. Vasaras pēcpusdienā, kārtējo reizi skrienot, pēkšņi zaudējis samaņu, nokritis. Par laimi, garām, no izsaukuma atgriežoties, brauca ātrās palīdzības brigāde un apkārtējie cilvēki to apturēja. Tika konstatēta kambaru fibrilācija un veikta defibrilācija. Pacientam atrodoties stacionārā, pierakstīja 12 novadījumu EKG, kas radīja aizdomas par kreisā kambara hipertrofiju.
Veicot ehokardiogrāfiju, konstatēja cirkulāru kreisā kambara hipertrofiju līdz 19 mm bez izejas trakta obstrukcijas. Tika sākta beta-adrenoblokatoru terapija, kategoriski aizliegta sportošana, pēc nedēļas pacientam implantēja ICD. Divu gadu laikā ICD nebija veicis ārstniecisko iejaukšanos, bet bija reģistrējis vairākus īslaicīgus kambaru tahikardijas paroksizmus. Ģenētiskā izmeklēšanā konstatēja divas ar HKMP saistītas mutācijas miozīna saistīšanas proteīna C gēnā (MYBPC3). Arī pacienta tēvam tika konstatētas tādas pašas ģenētiskās izmaiņas, taču viņam nav bijušas PKN epizodes, bet ehokardiogrammā vizualizēja mērenu KK hipertrofiju līdz 15 mm. Pacienta brālim izmaiņas gēnos netika diagnosticētas.
Dilatācijas kardiomiopātija (DKMP)
Par dilatācijas kardiomiopātiju sauc patoloģisku stāvokli, ko raksturo paplašināts kreisā kambara dobums un ievērojams sistoliskās funkcijas pasliktinājums, t.i., kreisā kambara izsviedes frakcija
Primārās un sekundārās DKMP
Primāras DKMP iemesls ir pārmantots ģenētisks defekts. Sekundāras - kāda cita slimība, piemēram, miokarda bojājums ar toksiskiem, metaboliskiem vai infekcioziem aģentiem, KSS, saistaudu saslimšanas utt. [7]
Salīdzinot ar HKMP, primāra DKMP ir retāka saslimšana, gadā tā tiek diagnosticēta aptuveni 20 no 100 000 iedzīvotāju.
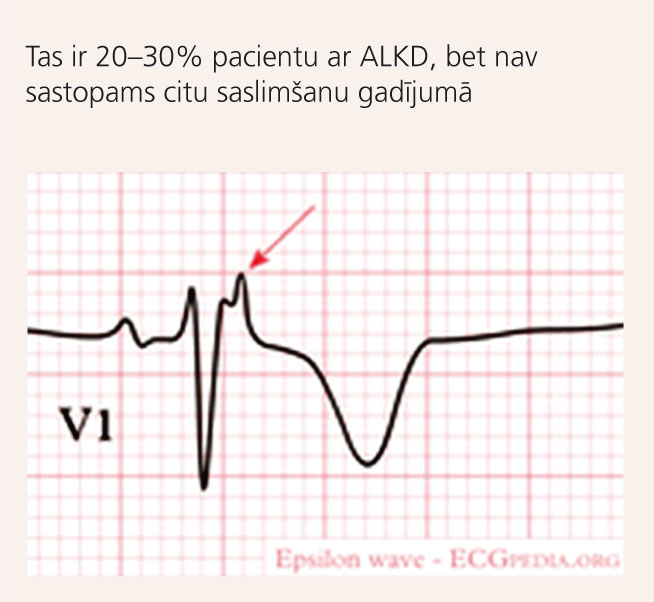
5. attēls
ALKD raksturīga pazīme EKG – “epsilona” vilnis
Primāras DKMP gadījumos ģenētiskie defekti skar dažādas sirds muskuļu olbaltumvielas (visbiežāk lamīnu A/C, bet var skart arī desmīnu, aktīnu, troponīnu T u.c.). Pašlaik vairāk nekā pusei gadījumu ģenētiskus iemeslus nav iespējams konstatēt, arī pirmās pakāpes radiniekiem ne. Šādās situācijās izmanto terminu "idiopātiska DKMP".
Ģenētisku iemeslu dēļ miokarda šķiedras paliek vājākas. Daļa no kardiomiocītiem iet bojā. Starp novājinātajām šķiedrām palielinās starpšķiedru vielas daudzums, notiek miokarda aizvietošana ar fibrotiskiem audiem. Rezultātā sirds dobumi dilatējas. Arī šajā gadījumā (līdzīgi HKMP) veidojas perēkļi ar traucētu elektriskā impulsa pārvadi, kas predisponē sirds ritma traucējumu attīstībai.
DKMP raksturīgas arī īpašas kambaru tahikardijas formas, kas saistītas ar Hisa kūlīša labo un kreiso zaru piedalīšanos jeb t.s. "fascikulāras tahikardijas". [8]
Atšķirībā no HKMP pie DKMP slimība no sākuma parasti izpaužas ar hroniskas sirds mazspējas radītiem simptomiem, vēlāk tiem pievienojas sirds ritma traucējumi. Būtisks fakts, ka pacientiem ar DKMP ļoti bieži vērojamas arī supraventrikulāras aritmijas, īpaši priekškambaru mirdzaritmija.
Diagnostika
Būtiskākais izmeklējums, kas apstiprina diagnozi, ir ehokardiogrāfija. Veicot EKG šiem pacientiem, var saskatīt zemas voltāžas QRS kompleksus, bieži intraventrikulārus vadīšanas traucējumus (Hisa kūlīša kreisā zara blokādi), sinusa tahikardiju, ļoti bieži ātriju fibrilācijas, nespecifiskas ST segmenta depresijas ar zemas voltāžas vai invertētiem T zobiem. [4]
Ārstēšana
Ieteikumi DKMP pacientu ārstēšanai:
- noskaidrot iespējamo iemeslu, t.sk. izmeklējot pacienta radiniekus;
- asimptomātiskiem pacientiem regulāri jāveic ehokardiogrāfiskā izmeklēšana. Atbilstīgi kreisā kambara dilatācijas pakāpei tā būtu jāveic reizi 6-24mēnešos;
- parādoties hroniskas sirds mazspējas simptomiem vai ritma traucējumiem, pacienta ārstēšana un tālākā taktika jāsaskaņo ar speciālistu, lai lemtu par:
- medikamentozu terapiju: atbilstīgi situācijai lieto beta-adrenoblokatorus, angiotenzīna konvertējoša enzīma inhibitorus, aldosterona antagonistus, diurētiķus, sirds glikozīdus, vazodilatatorus,
- implantējamu ierīci: pastāvīgu biventrikulāru elektrokardiostimulatoru ar vai bez kardiovertera-defibrilatora funkcijas (CRT vai CRT-D) implantāciju.
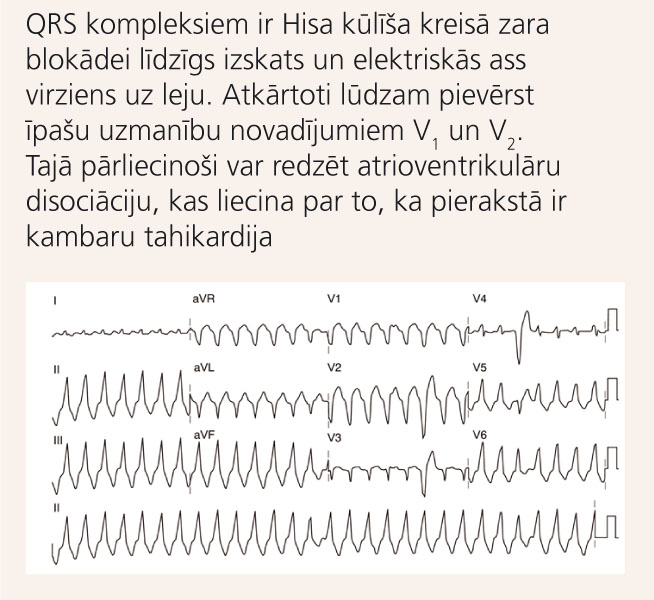
6. attēls
EKG ar ALKD raksturīgu monomorfu kambaru tahikardiju
Aritmogēna labā kambara displāzija (ALKD)
Salīdzinot ar divām iepriekšējām patoloģijām, pētījumi par ALKD tiek veikti relatīvi nesen. Pēdējos 20 gados par šo slimību ir izdevies noskaidrot ļoti daudz, bet vēl joprojām ALKD diagnoze ir viena no sarežģītākajām mūsdienu kardioloģijā. Visbūtiskākais ir fakts, ka šī slimība ļoti bieži izpaužas kā PKN gados jauniem cilvēkiem, īpaši tiem, kas nodarbojas ar sportu.
ALKD ir iedzimta saslimšana, kuras iemesls visbiežāk ir patoloģiskas pārmaiņas šūnu kontaktu - desmosomu uzbūvē. Šūnu kontaktu disfunkcija rada iesaistīto miokarda šūnu bojāeju un aizvietošanu ar fibrotiskiem audiem un taukaudiem (skat. 4. attēlu). Šādas izmaiņas visbiežāk novēro labajā kambarī, t.s. displāzijas trīsstūrī - labā kambara subtrikuspidālajā zonā, galotnē un izplūdes traktā. Slimības vēlīnās fāzēs var iesaistīties arī kreisais kambaris, un tas ir saistīts ar sliktu prognozi.
ALKD ļoti bieži noris asimptomātiski. Pacientiem, kam slimība norit ar simptomiem, pat 60-80% gadījumu tās pirmā pazīme ir pārsitieni, ģībonis vai PKN fiziskas slodzes laikā. Tas var būt slimības pirmais simptoms, kam pievērš uzmanību. Tomēr ALKD raksturīgās pazīmes parasti var konstatēt pacientiem arī asimptomatiskajā periodā, taču tās bieži vien kļūdaini interpretē kā nenozīmīgas pārmaiņas vai normas variantu.
Diagnostika
EKG pazīmes, kas varētu liecināt par ALKD:
- invertētie Tviļņi labajos prekardiālos krūšu novadījumos V1-V3, ja nav Hisa kūlīša labās kājiņas blokādes (pacientiem pēc 12gadu vecuma);
- epsilona vilnis vai lokāla QRS kompleksa pagarināšanās virs 110msek. V1-V3 krūšu novadījumos (skat. 5.attēlu);
- nepilna Hisa kūlīša labā zara blokāde.
Veicot Holtera monitorēšanu, pacientiem ar ALKD var novērot biežas kambaru ekstrasistoles (vairāk par 1000 24 stundu laikā) vai pat kambaru tahikardijas paroksizmus ar QRS kompleksiem ar Hisa kūlīša kreisā zara pilnas blokādes morfoloģiju (skat. 6. attēlu). [9]
Līdzīga QRS kompleksu morfoloģija ir arī no kambaru izplūdes traktu izejošām idiopātiskām kambaru ekstrasistolēm un kambaru tahikardijām, kas ir biežākā diferenciāldiagnoze. Atšķirība no ALKD šie ritma traucējumi parasti ir labdabīgi un nav saistīti ar PKN.
Atrodot ALKD raksturīgas izmaiņas EKG, tālāka izmeklēšana saistīta ar EhoKG vai citu vizualizācijas metodi - magnētisko rezonansi, datortomogrāfiju ar kontrastvielu, ventrikulogrāfiju. Ja kādā no tām tiek konstatēti labā kambara kustību vai funkciju traucējumi, pacients jāsūta uz konsultāciju pie speciālista.
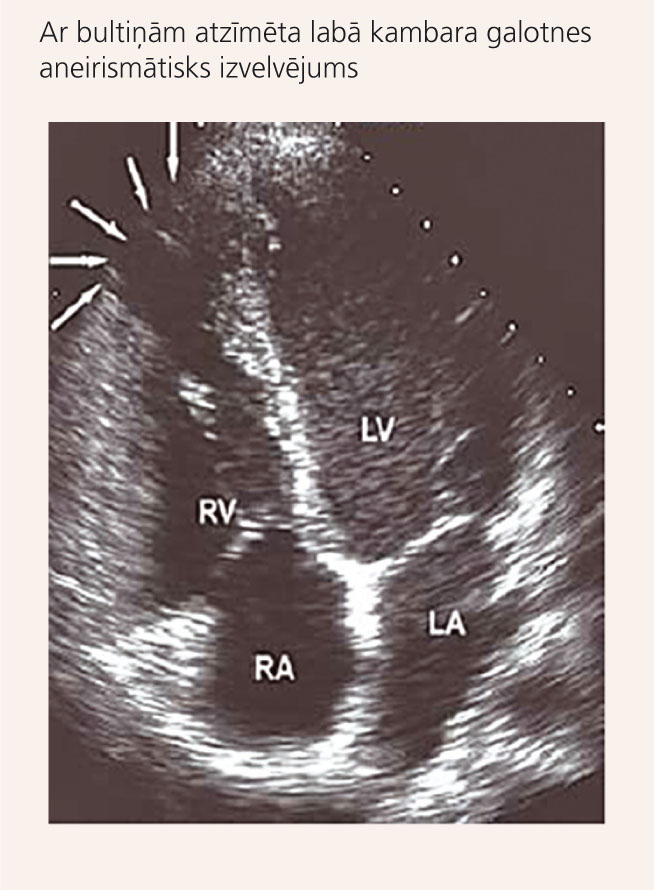
7. attēls
Ehokardiogrāfiska atrade pacientei ar ALKD
Ārstēšana
ALKD ārstēšana ir diezgan problemātiska. Grūtākais jautājums ir par ārstēšanas sākšanas nepieciešamību asimptomātiskiem pacientiem un simptomātiska pacienta radiniekiem, kam ir noteiktas slimībai raksturīgās pazīmes. Ārstēšanas iespējas:
- farmakoterapija: sirds ritma traucējumu korekcijai lietojami beta-adrenoblokatori un citi antiaritmiskie līdzekļi, īpaši Sotaloli hydrochloridum. Parādoties sirds mazspējai, tā jāārstē līdzīgi citiem pacientiem;
- implantējamie kardioverteri-defibrilatori.
Gadījums Latvijā
Sieviete, 28 gadus veca, pēc profesijas grāmatvede, jau 5-7 gadus sūdzējās par īslaicīgām sinkopēm. Bezsamaņas parasti ir pie fiziskas slodzes. Slimība traucēja nodarboties ar basketbolu, bieži spēles laikā reibusi galva, dažreiz arī zaudēja samaņu. Veicot Holtera monitorēšanu, tika konstatētas apmēram 8000 (9% no visiem QRS kompleksiem) kambaru ekstrasistoles ar Hisa kūlīša kreisā zara pilnas blokādes morfoloģiju. Pēc aritmologa ieteikuma veikta ehokardiogrāfija, kur konstatēja aneirismu labā kambara galotnē (skat. 7. attēlu). Veiktajās ģenētiskajās analīzēs pacientei konstatēta ar ALKD saistīta mutācija desmokolīna-2 gēnā (DSC2). Ņemot vērā ģīboņus un augstu PKN risku, pacientei tika piedāvāta ICD implantācija, paciente no tās šobrīd ir atteikusies, bet regulāri lieto beta-adrenoblokatorus.
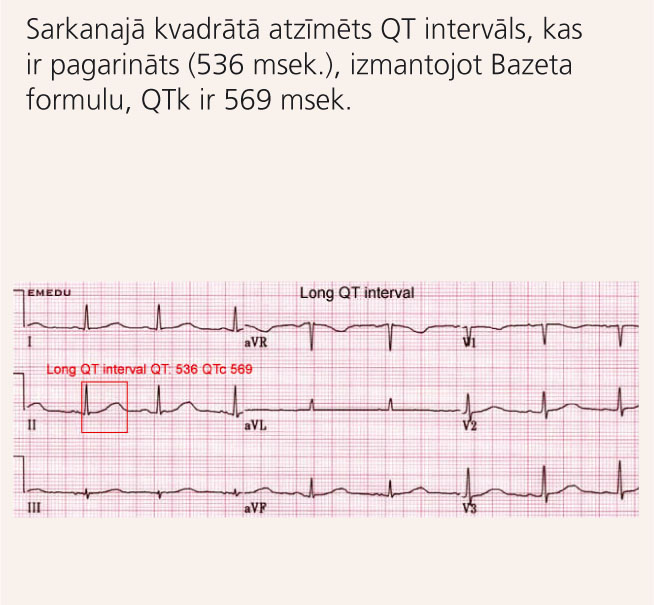
8. attēls
EKG pacientam ar iedzimtu pagarināta QT intervāla sindromu
Iedzimts pagarināts QT intervāla sindroms
Šā sindroma pamatā ir pārmantots kardiomiocītu jonu kanālu proteīnu defekts, kura dēļ pagarinās kambaru repolarizācija. Tādējādi rodas nosacījumi ventrikulāru aritmiju attīstībai. Slimībai raksturīgas polimorfas kambaru tahikardijas (torsades de pointes), kas var pāriet kambaru fibrilācijā. Sinkopes un PKN pirmā epizode parasti notiek pusaudžu gados, bet var būt arī agrā bērnībā. Pēc 40 gadu vecuma slimība sākas reti.
Svarīgi ir atšķirt iedzimtu pagarinātu QT sindromu no klīniskiem stāvokļiem, ko raksturo pagarinātais QTk intervāls jeb t.s. iegūtas QTk intervāla pagarināšanas.
Iegūta QTk pagarināšana vērojama, ja ir izraisošs aģents, piemēram, ja lieto dažādus medikamentus, vai miokarda infarkta akūtajā fāzē. Mazinoties šo aģentu ietekmei, QTk intervāls normalizējas. Būtiski ir atcerēties: pagarinoties QTk intervālam, pacientiem draud tās pašas bīstamības, kādas iedzimta pagarināta QT intervāla sindroma gadījumā.
QTk intervāla pagarināšanos izraisa: [4]
- dažādi medikamenti:
- antiaritmiskie līdzekļi- kordarons, sotalols, hinidīnsu.c.,
- psihiatrijā izmantojamie medikamenti- amitriptilīns, citaloprams, fluoksetīns, venlaflaksīns, haloperidols, risperidons, kvetiapīnsu.c.,
- antibiotikas- eritromicīns, klaritromicīns, ciprofloksacīnsu.c.,
- antihistamīna preparāti, īpaši pirmās paaudzes, piemēram, cimetidīns,
- citi medikamenti;
- elektrolītu traucējumi:
- hipokaliēmija,
- hipomagniēmija,
- hipokalciēmija;
- slimības- miokarda infarkts (īpaši akūtajā fāzē), miokardīts, hipotireozeu.c.
Diagnostika
Visvērtīgākais izmeklējums ir EKG [1] (skat. 8. attēlu). Galvenais riska faktors sinkopēm un PKN ir t.s. koriģētais QT intervāls (QTk jeb QTc). Tā noteikšanai visbiežāk izmanto Bazeta formulu:
QTk = QT / kvadrātsakne no R-R intervāla.
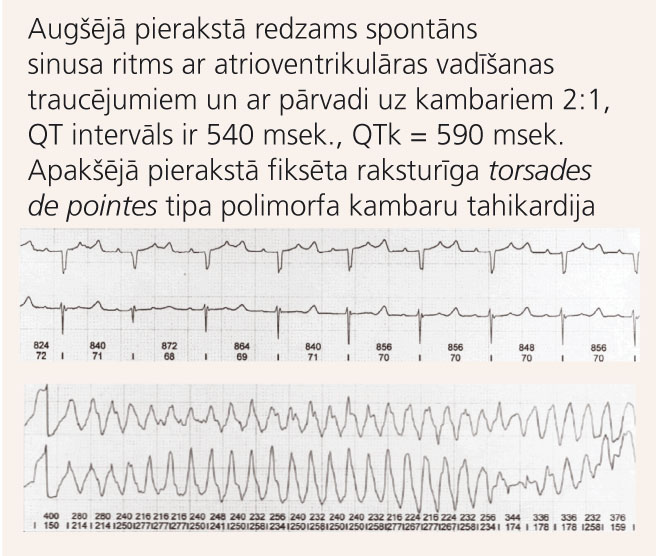
9. attēls
Holtera monitorēšanas pieraksts pacientei ar iedzimtu garā QT intervāla sindromu
QTk un QT var izteikt sekundēs vai milisekundēs, bet R-R intervālu izsaka sekundēs.
Augšējā norma QTk intervālam ir zem 460 msek. sievietēm un zem 440 msek. vīriešiem. Taču viss nav tik viennozīmīgi! Trīs problēmas, kas padara šīs slimības diagnostiku sarežģītāku:
- aprēķinu ticamība ievērojami mazinās, ja pacientam ir izteikta bradikardija vai tahikardija;
- veselā populācijā 2,5% gadījumu ir pagarināts QTk intervāls, savukārt 10-15% gara QT intervāla sindroma gadījumos QTk intervāls ir normāls;
- nosakot QTk intervālu, mērījumos nedrīkst iekļaut Uzobu, jo tad var panākt "pseidopozitīvu" rezultātu.
Ārstēšana
Pacientiem ar iedzimtu pagarinātu QT intervālu būtu jāizvairās no to medikamentu lietošanas, kas pagarina QTk intervālu.
Pacienti ar aizdomām par pagarināta QT intervāla sindromu būtu jānosūta pie speciālista uz konsultāciju tālākai izmeklēšanai un ārstēšanai. Apsveramie pasākumi:
- pacienta un viņa radinieku izglītošana. Pacientu informētībai ir liela nozīme jeb kuras slimības gadījumā, t.sk. to sli mī bu, ko esam apskatījuši, taču iedzimta pagarināta QT intervāla sindroma gadījumā tai ir īpaši liela nozīme. Piemēram, pacientiem obligāti jābūt informētiem par aizliegtajiem medikamentiem un par citiem QT intervālu pagarinošiem stāvokļiem;
- ārstēšanas būtiskākais līdzeklis ir beta-adrenoblokatori. Reizēm jālieto diezgan lielas devas, dažreiz pacientiem jāimplantē pastāvīgais elektrokardiostimulators;
10. attēls
EKG pacientam ar Brugada sindromu
- implantējamie kardioverteri-defibrilatori.
Gadījums Latvijā
Astoņus mēnešus vecai meitenītei kopš dzimšanas vecāki periodiski novēroja apnojas un akrocianozes epizodes. Veikta Holtera monitorēšana, kur konstatēts garš QT intervāls un atrioventrikulāras vadīšanas traucējumi (skat. 9. attēlu). Pacientei veikta pastāvīga divkameru elektrokardiostimulatora implantācija un sākta terapija ar beta-adrenoblokatoriem. Veiktajās ģenētiskajās analīzēs konstatēja izmaiņas kālija kanālu veidojošā proteīna gēnā (KCNH2), kas ir saistītas ar iedzimtu pagarināta QT intervāla sindromu. Izmeklējot bērna radiniekus, arī tēvam tika apstiprināta šā gēna izmaiņas, bet QT intervāls EKG ir normāls, viņam nebija arī datu par sirds ritma traucējumiem - asimptomātiska gēna nēsāšana.
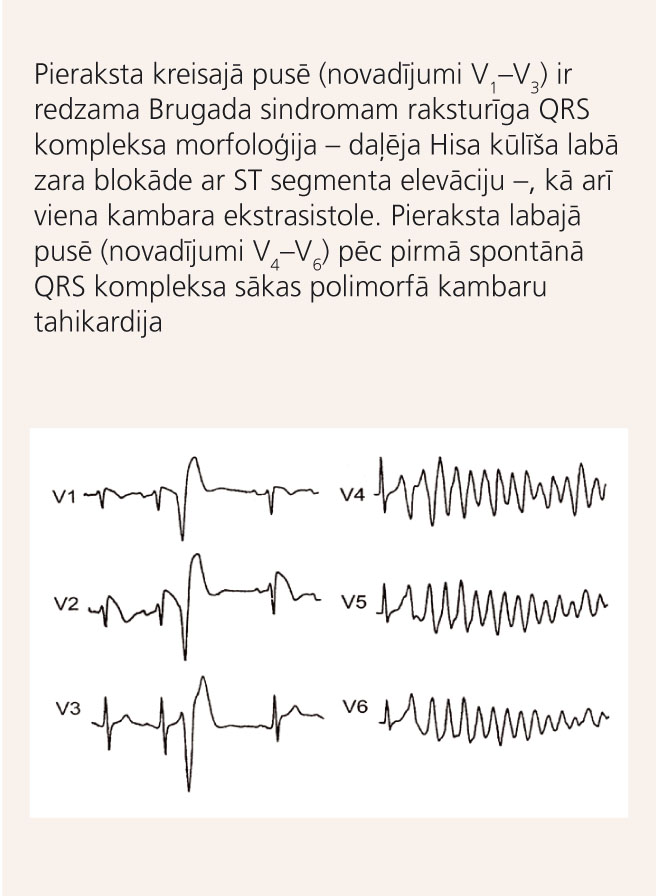
Brugada sindroms
Brugada sindroms ir iedzimta saslimšana, ko EKG raksturo Hisa kūlīša labās kājiņas blokādes aina ar ST segmenta elevāciju labajos prekardiālajos novadījumos un PKN. [1]
Līdzīgi iedzimtam gara QT intervāla sindromam arī šīs slimības pamatā ir izmaiņas kardiomiocītu jonu kanālu darbībā. Brugada sindroma gadījumā visbiežāk defekts skar gēnu, kas kodē nātrija jonu kanālu veidojošu proteīnu. [1]
Vēl viena līdzība ar iedzimtu pagarinātu QT intervāla sindromu - abām šīm slimībām izmaiņas konstatējamas tikai sirds elektriskajā sistēmā. Pacientiem ar iedzimtu gara QT intervāla sindromu vai Brugada sindromu nav izmaiņu, ko varētu noteikt ar vizualizācijas metodēm.
Vienīgā Brugada sindroma klīniskā izpausme ir sinkopes polimorfu kambaru tahikardiju dēļ (skat. 10. attēlu) vai pat PKN. Saistība ar fizisku slodzi šim sindromam nav raksturīga, ritma traucējumu epizodes parasti notiek naktīs vai agrās rīta stundās.
Diagnostika
Diagnozes pierādīšanai ļoti vērtīga ir elektrokardiogramma. Brugada sindromam ir trīs EKG tipi (skat. 11. attēlu). [10]Izmaiņas EKG pastiprinās dažādos apstākļos, piemēram, lietojot medikamentus, kā arī tad, ja ir paaugstināta temperatūra u.c. Adrenerģiska stimulācija samazina ST segmentu elevāciju, bet vagāls uzbudinājums to palielina. Tāpēc biežāk nāves gadījumi tiek novēroti miera stāvoklī, miegā un nevis pie slodzes. [12]
Līdzīgi kā pie iedzimta pagarināta QT intervāla sindroma arī pie Brugada sindroma ir medikamenti, kuru lietošana varētu provocēt izmaiņas EKG un sirds ritma traucējumus (piemēram, flekainīns, prokaīnamīds, propafenons, amitriptilīns, fluoksetīns, propofols, karbamazepīns, amiodarons u.c). Tāpēc pacientiem ar pierādītu Brugada sindromu šos preparātus neiesaka lietot.
Ārstēšana
Rodoties aizdomām par šo sindromu, pacients jāsūta pie speciālista tālākai izmeklēšanai, diagnozes precizēšanai un ārstēšanai. Iespējamās darbības:
- pacientiem ar aizdomām par Brugada sindromu vajadzētu veikt medikamentu provokācijas testus;
- ja pacientam konstatē izmaiņas, obligāti jāpārbauda viņa radinieki;
- nav iesakāma un pat bīstama varētu būt beta-adrenoblokatoru un citu antiaritmisku līdzekļu lietošana;
- augsta riska pacientiem indicēta implantējama kardiovertera-defibrilatora implantācija.
Gadījums Latvijā
43 gadus veca sieviete, pēc profesijas sekretāre, sūdzējās par īslaicīgām bezsamaņas epizodēm vairāku gadu garumā. Atslēgšanās epizodes dēļ vienu reizi iekļuvusi nelielā satiksmes negadījumā. Aizdomās par neiroloģisku saslimšanu pacientei veikta elektroencefalogramma un brahiocefālo asinsvadu izmeklēšana, kas patoloģiju neuzrādīja. Standarta 12 novadījumu EKG uzrādīja pazīmes, kas lika aizdomāties par Brugada sindromu, tad veikta EKG ar farmokoloģisku provokācijas testu (skat. 12. attēlu). Augsta PKN riska dēļ pacientei implantēja ICD.
Pacientes dēlam un māsai EKG arī konstatētas Brugada sindromam raksturīgas pārmaiņas. Ģenētiskās analīzes nosūtītas uz laboratoriju, tiek gaidīta atbilde.
Kateholamīnerģiska polimorfa ventrikulāra tahikardija (KPVT)
Šī ir iedzimta un ļoti ļaundabīga, bet reta aritmogēna saslimšana. To raksturo morfoloģiski normāla sirds, bet polimorfas kambaru tahikardijas emocionālu pārdzīvojumu vai fiziskas slodzes laikā.
Slimības pamatā visbiežāk ir izmaiņas kalcija kanālu jeb t.s. rianodīna receptoru veidojošā proteīna kodējošos gēnos. Rezultātā daļa no sarkoplazmatiskā tīkla kalcija joniem iekļūst miocītu citosolā. Tāpēc uz it kā parastiem stimuliem sirds atbild ievērojami spēcīgāk. Piemēram, emocionāli pārdzīvojot vai nodarbojoties ar sportu, parādās ne tikai sinusa tahikardija, bet arī atsevišķas kambaru ekstrasistoles vai arī kambaru tahikardijas, kas diezgan bieži var pāriet kambaru fibrilācijā.
Klīniski raksturīgākā KPVT pazīme ir epizodiskas sinkopes vai PKN, kas parādās fiziskas slodzes laikā vai emocionālu pārdzīvojumu apstākļos. Bezsamaņu iemesls ir kambaru tahikardijas, kas diezgan ātri var pāriet kambaru fibrilācijā (skat. 13. attēlu). Vidējais vecums, kad parādās simptomi, ir 7-11 gadi. Retāk slimības sākums ir bērniem pirms 3 gadu vecuma vai arī vecākiem cilvēkiem (pusaudžiem). [3]
Diagnostika
Miera stāvoklī KPVT pacientiem EKG ir bez būtiskām izmaiņām, dažreiz var novērot sinusa bradikardiju un U vilni. [13]Diagnozes pierādīšanai ieteicams veikt Holtera monitorēšanu un slodzes testu. Tipiskākā atradne pie KPVT ir atsevišķas kambaru ekstrasistoles vai kambaru tahikardijas, kas saistītas ar fizisku slodzi vai emocionāliem pārdzīvojumiem. Aizdomām apstiprinoties un ņemot vērā slimības ļaundabīgu gaitu, pacienti ar aizdomām par KPVT jāsūta pie speciālista.
Ārstēšana
Iespējama ārstēšana:
- pacientiem ar apstiprinātu diagnozi kategoriski aizliegts nodarboties ar sportu vai veikt smagus fiziskus darbus;
- izvēles medikaments pacientiem ar KPVT ir beta-adrenoblokatori, t.sk. slimības akūtajā periodā. Dažreiz jālieto diezgan augstas devas, ja pacients to panes;
- alternatīva varētu būt ne-dihidropiredīnu grupas kalcija kanālu blokatori. Datu par to lietošanas efektivitāti ir maz;
- pacientiem ar augstu PKN risku un biežām kambaru tahikardijas epizodēm jāimplantē ICD.