Iekaisuma process
Iekaisums tiek traktēts kā kompleksa organisma reakcija uz mikroorganismiem, ķīmiskām vielām vai fizikāliem aģentiem vaskularizētos audos, kuras rezultātā notiek cirkulējošo leikocītu, saistaudu šūnu, ektracelulāro komponentu (kolagēna, elastīna), glikoproteīnu (fibronektīna, laminīna, proteoglikānu) infiltrācija šajos audos. [4] Četras iekaisuma pazīmes jau pirmajā gadsimtā formulēja Celzs (Celsus): tumor, rubor, dolor, color. Piekto pazīmi - functio laesa - vēlāk pievienoja R. Virhovs.
18. gadsimta beigās skotu ķirurgs Džons Hanters (John Hunter), balstoties uz saviem novērojumiem, secināja, ka iekaisums nav slimība, bet nespecifiska reakcija, kurai ir dziedinoša ietekme uz organismu. Vēlākos gados tiek ļoti aktīvi pētīti iekaisuma procesi, un 1882. gadā krievu zinātnieks Iļja Mečņikovs atklāj fagocitozi un secina, ka iekaisuma mērķis ir aizgādāt fagocītus uz bojājuma vietu, lai tie aprītu tur esošās baktērijas. Tika likti pamati celulārajai iekaisuma teorijai. Nedaudz vēlāk Pauls Ērlihs (P. Erlich) atklāja faktorus serumā (mūsdienu izpratnē - antivielas), kas spēj cīnīties pret mikroorganismiem un formulēja humorālās iekaisuma teorijas pamatus. 1908. gadā abi zinātnieki saņēma Nobela prēmiju par ieguldījumu iekaisuma procesu pētīšanā.
Laikam ritot, pasaules zinātnieki formulēja vienkāršākas un sarežģītākas iekaisuma definīcijas. Viena no saprotamākajām un lakoniskākajām šķiet sekojošā: „Iekaisums ir organisma aizsargreakcija - dzīvo audu reakcija uz bojājumu, kas izraisa asinsvadu, asins formelementu un saistaudu izmaiņas, kas vērstas uz iekaisuma izraisītāja elimināciju un bojāto audu atjaunošanu."
Pēdējo gadu pētījumi imunoloģijā mudinājuši Amerikas Nacionālā Vēža institūta Audzēju bioloģijas izpētes grupu 2006. gadā formulēt laikmetīgāku iekaisuma definīciju: „Iekaisums ir komplicēta mijiedarbība starp imūnšūnām un šķīstošajiem mediatoriem, kas veidojas kā atbildes reakcija uz audu bojājumu traumas, infekcijas, toksisku aģentu vai autoimūnu procesu dēļ un kuras rezultāts ir lokāla šūnu proliferācija un bojāto audu atjaunošana."
Iekaisuma reakcija ir nespecifiska imūnreakcija, kuras norises galvenos etapus varētu aprakstīt ļoti shematiski, par piemēru izvēloties iekaisuma reakciju brūcē:
- baktērija vai cits patogēns iekļūst organismā caur brūci,
- trombocīti izdala asins recēšanu veicinošus proteīnus brūces vietā,
- tuklās šūnas izdala faktorus, kas regulē vazodilatāciju un vazokonstrikciju; pastiprinās asins un tās formelementu pieplūde bojājuma vietai,
- neitrofili sekretē bioloģiski aktīvas vielas, kas nonāvē un degradē patogēnus,
- neitrofili un makrofāgi fagocitē patogēnus,
- makrofāgi sekretē citokīnus, kas piesaista imūnšūnas un aktivē procesus, kuri saistās ar audu reģenerāciju,
- iekaisuma atbilde turpinās tik ilgi, līdz patogēns ir eliminēts un brūce - sadzijusi [5].
Iekaisuma veidošanās shēma parādīta 1. attēlā.
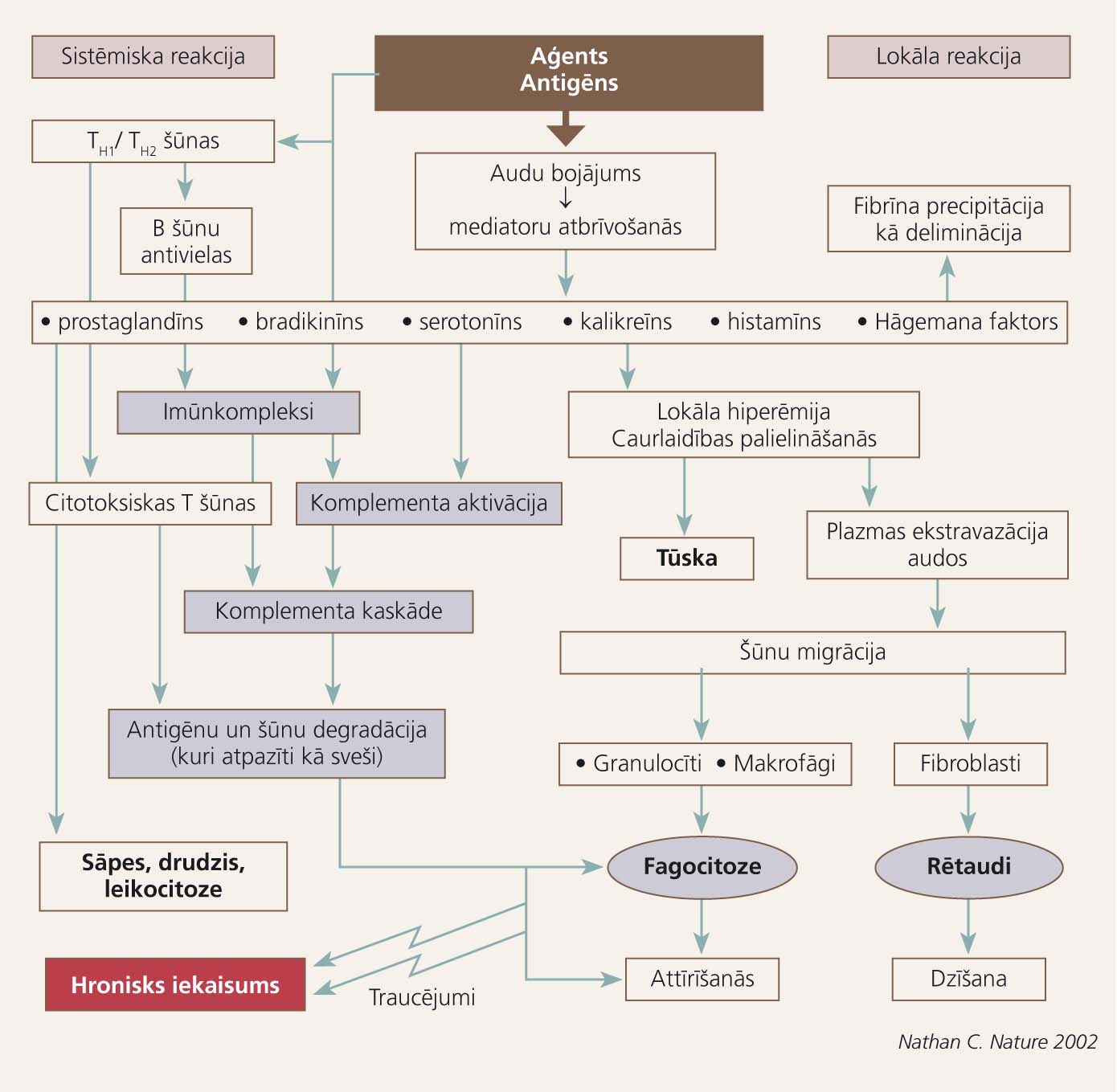
1. attēls
Vienkāršota iekaisuma veidošanās shēma
Akūts iekaisums
Akūta iekaisuma laikā notiek strukturālas izmaiņas asinsvadu sienās un mikrovidē ap asinsvadiem: plazmas proteīnu un leikocītu ekstravazācija, migrācija uz bojājuma vietu un fagocitoze.
Akūta iekaisuma iznākums ir procesa uzsūkšanās (izveseļošanās), fibroze vai hronisks iekaisums.
Hronisks iekaisums
Hronisks iekaisums ir ieilgusi iekaisuma reakcija, kad aktīvs iekaisums, audu destrukcija un bojāto audu atjaunošanās notiek vienlaicīgi.
Hronisks iekaisums var ne tikai sekot akūtam, bet arī izpausties kā neaktīva, asimptomātiska reakcija uz kairinātāju persistējošas infekcijas, ilgstošas toksisku aģentu iedarbības vai autoimūnu procesu dēļ. Hroniska iekaisuma perēklis raksturojas ar mononukleāro šūnu - makrofāgu, limfocītu, plazmas šūnu -, nevis neitrofilu infiltrāciju. Iekaisuma perēklī notiek šūnu nekroze un bojātie audi hroniska procesa gadījumā angioģenēzes un fibrozes rezultātā tiek aizstāti ar saistaudiem.
Hroniska iekaisuma laikā, atbrīvojoties virknei bioloģiski aktīvu vielu, tiek izjaukts līdzsvars starp šūnu proliferāciju un apoptozi, kas izraisa nekontrolētu šūnu dalīšanos un transformētu šūnu vairošanās iespēju. Aktivēti makrofāgi un limfocīti ir lielākais proinflamatoro citokīnu, augšanas faktoru un angioģenēzes stimulatoru avots. Citokīni, hemokīni un augšanas faktori mijiedarbojas ar specifiskiem receptoriem uz šūnas virsmas, kas dod signālu šūnu proliferācijā iesaistītiem gēniem un ietekmē ļaundabīgi izmainīto šūnu dzīvotspēju, neoangioģenēzi un audzēja šūnu migrāciju apkārtējos audos. [4]
Hroniska iekaisuma laikā noritošā fagocitozes procesā radušies metabolisma produkti veicina pastiprinātu brīvo radikāļu veidošanos, kas spēj bojāt DNS, lipoproteīnus un šūnu membrānu. Iekaisuma procesā iesaistītās šūnas izdala virkni bioloģiski aktīvu vielu (prostaglandīnus, arahidonskābes metabolītus, leikotriēnus), kas spēj ietekmēt kanceroģenēzi. Aktivētu iekaisuma šūnu un stromas elementu radītajā mikrovidē ātrāk varētu noritēt proliferējošu cilmes šūnu neoplastiska transformācija un turpmāka invāzija apkārtējos audos. [6]
Dvoraks (Dvorak) audzēju nosauca par brūci, kas nedzīst. Zinātnieks uzsvēra atšķirības starp fizioloģisku, brūču dzīšanu pavadošu iekaisumu un tām norisēm organismā, kas saistās ar patoloģisku hronisku iekaisumu un veicina ļaundabīgu šūnu transformāciju, proliferāciju un invāziju. [7]
Hronisks iekaisums un ļaundabīga audzēja veidošanās
Hroniska iekaisuma loma tiek uzsvērta visos ļaundabīgā audzēja veidošanās posmos; hroniska iekaisuma gaitā notiekošie procesi ietekmē gan audzēja iniciāciju un progresiju, gan tā invāziju un metastazēšanos.
Kā jau iepriekš minēts, hroniska iekaisuma perēklī intensīvi darbojas makrofāgi, pastiprināti veidojas aktīvie skābekļa un slāpekļa savienojumi, kas spēj izraisīt epitēlijšūnu DNS bojājumus. Otrs faktors, kas sekmē audzēja iniciāciju, ir iekaisuma vietā koncentrējušos šūnu pastiprināta spēja izdalīt citokīnus, kas var inducēt vai pastiprināt proliferācijas signālus un veicināt to šūnu vairošanos, kurās varētu būt mutācijas. Audzēja iniciācijas vienkāršota izteiksme ir: DNS bojājums + pastiprināta proliferācija = audzēja aizsākums. [8; 9]
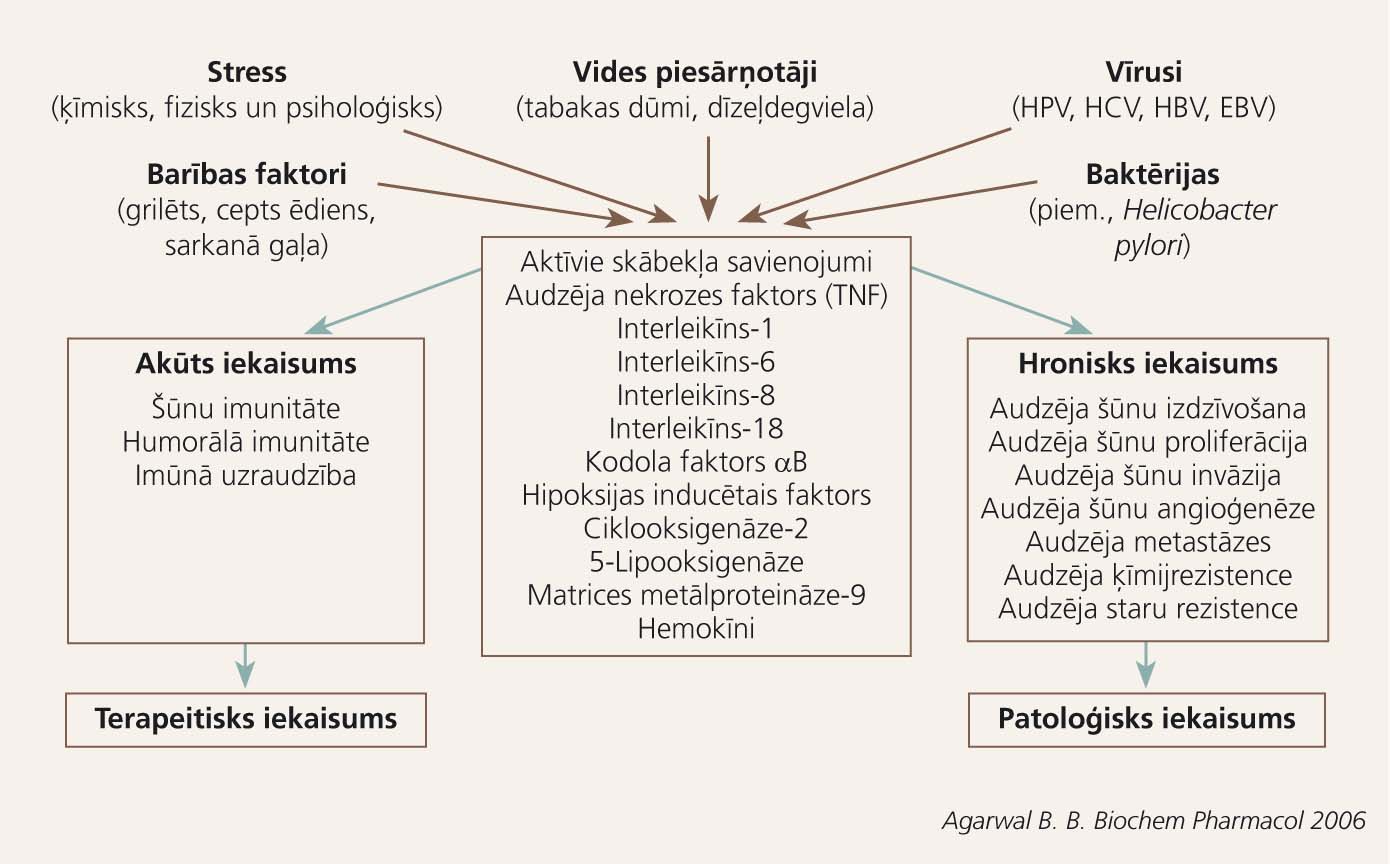
Turpmāko audzēja progresiju uz hroniska iekaisuma fona veicina vēža šūnu un imūnšūnu kompleksā mijiedarbība. Virkne bioloģiski aktīvo vielu - audzēja nekrozes faktors-alfa (TNF-a), aktīvie skābekļa savienojumi, IL-1, IL-6, IL-8, IL-18, nukleārais faktors kappa B (NFkB), hipoksijas inducētais faktors (HIF), ciklooksigenāze-2 (COX-2), matrices metālproteināze-9 (MMP-9) un citi - veicina ļaundabīgo šūnu izdzīvošanu un augšanu, kā arī angioģenēzi, kas vitāli nepieciešama audzēja progresijas fāzē. Daļa šo aktīvo substanču darbojas imūnsupresīvi, palīdzot audzējam izvairīties no imūnās kontroles. [10; 11]
Nākamajā audzēja attīstības - invāzijas un metastazēšanās - fāzē būtiska nozīme ir mijiedarbībai starp audzēja un epitēlija šūnām: invāzijas procesā ļaundabīgi transformētajām šūnām jāpārvar bazālā membrāna un asinsvadu endotēlija barjera. Šai procesā liela loma ir adhēzijas molekulām, kuru ekspresija hroniska iekaisuma mediatoru klātbūtnē pastiprinās kā uz endotēlija, tā arī uz audzēja šūnu virsmas. Šo šūnu savstarpējie kontakti kļūst ciešāki, un tas veicina audzēja invāziju. Hroniska iekaisuma vidē esošie citokīni un hemokīni stimulē audzēja šūnu migrāciju caur asinsvada sienu. Arī jau noformējušos metastāžu vietās iekaisums turpina veicināt to progresiju, iesaistoties transformējošam augšanas faktoram-beta (TGF-beta), kas ietekmē virkni bioloģisko procesu, kurus saista ar šūnu apoptozes regulāciju. [12]
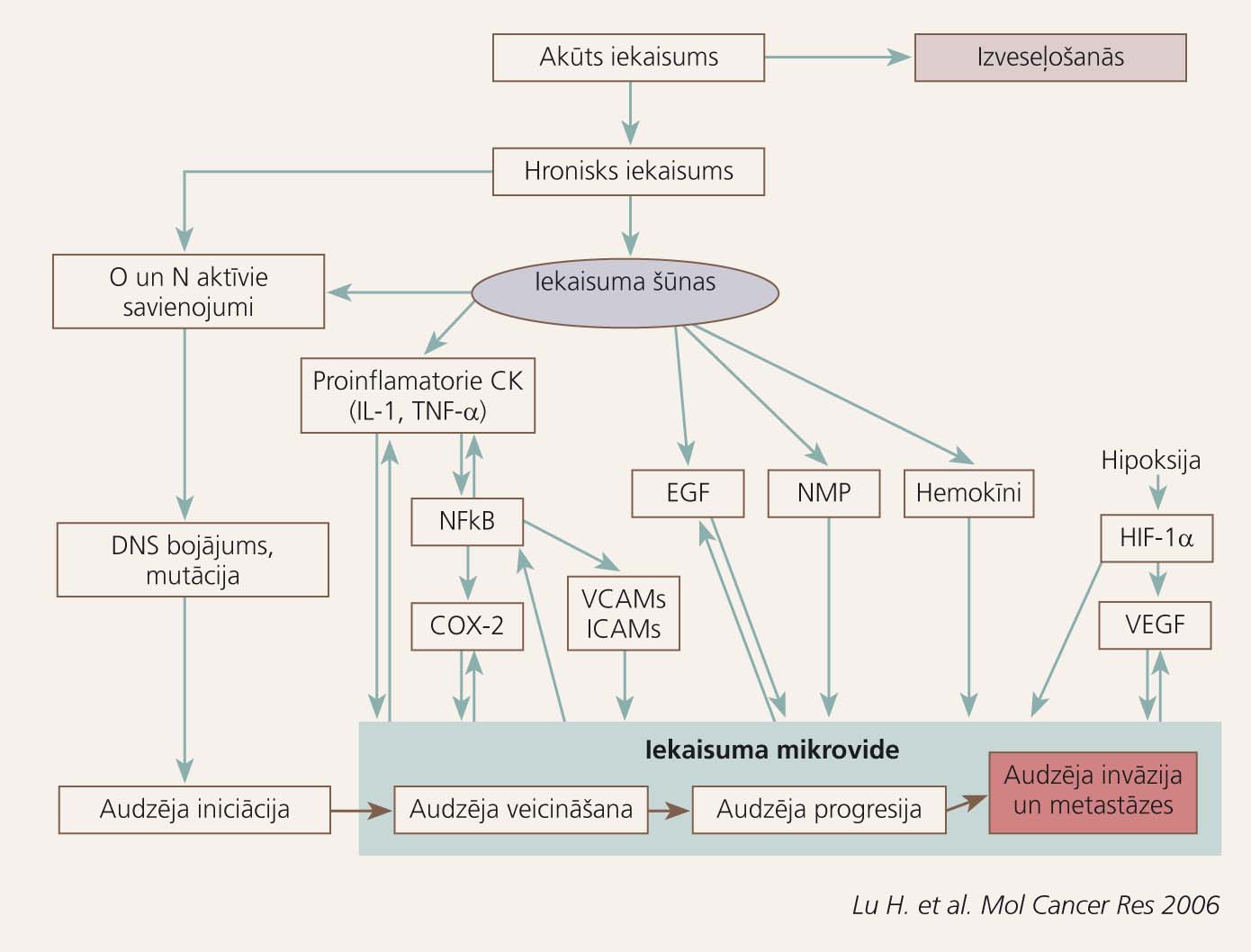
2. attēls
Infekciozie un ķīmiskie kairinātāji un audu bojājums
Iekaisuma mediatori saistībā ar kanceroģenēzes fāzēm 2. attēlā atspoguļo Virhova aprakstīto iekaisuma un ļaundabīga audzēja sakarību mūsdienu skatījumā. Bioloģiski aktīvās vielas, kas izdalās hroniska iekaisuma laikā, un to ietekme uz kanceroģenēzi apkopota 1. tabulā.
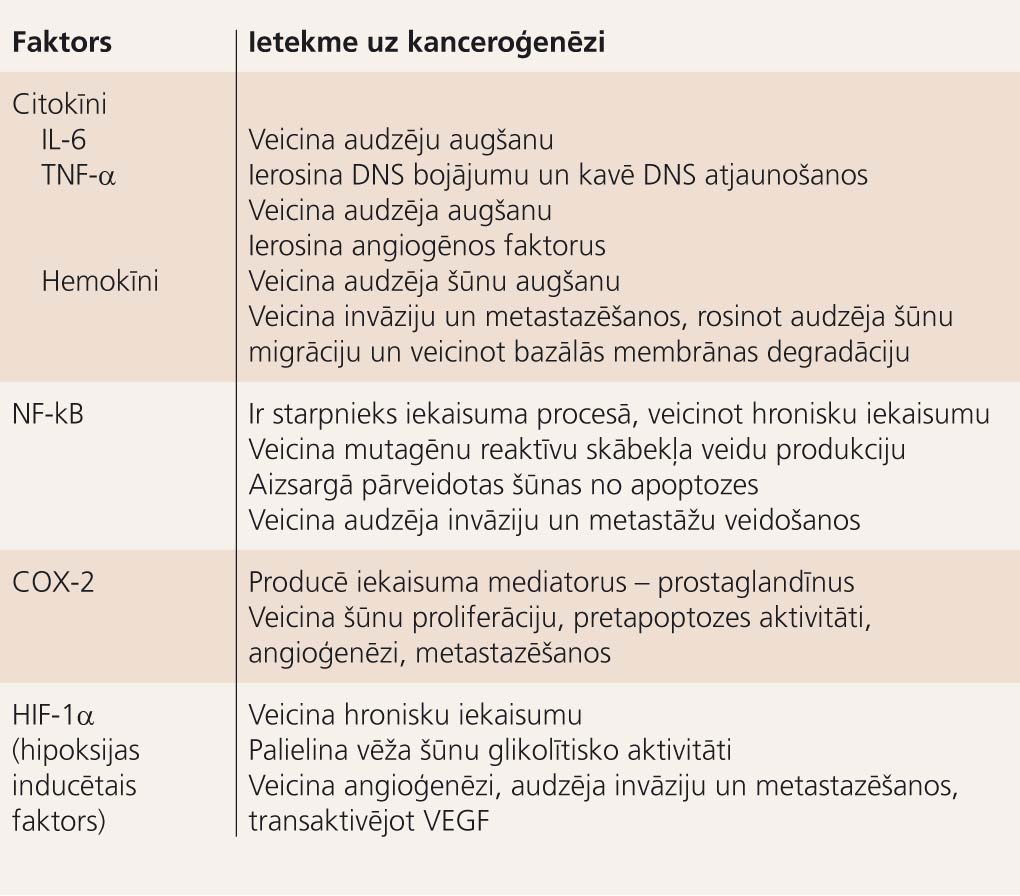
1. tabula
Bioloģiski aktīvās vielas, kas izdalās hroniska iekaisuma laikā, un to ietekme uz kanceroģenēzi
Ar hronisku iekaisumu saistīto gremošanas sistēmas orgānu ļaundabīgi audzēji
Gremošanas trakta orgānu iekaisums ir bieža parādība klīniskajā praksē, arī hroniski gadījumi nav retums, un, ņemot vērā iepriekš apskatītos procesus, kas notiek hroniska iekaisuma perēklī, pastāv varbūtība, ka iekaisums pāriet ļaundabīgā audzējā. Jebkura gremošanas trakta orgāna neizārstēts iekaisums var pārtapt audzējā (2. tabula). Turpmāk aplūkotie piemēri vienīgi ilustrēs hroniska iekaisuma saistību ar ļaundabīgo audzēju kādā konkrētā orgānā, mainoties iekaisuma cēlonim un saglabājoties līdzīgām patoģenētiskajām norisēm.
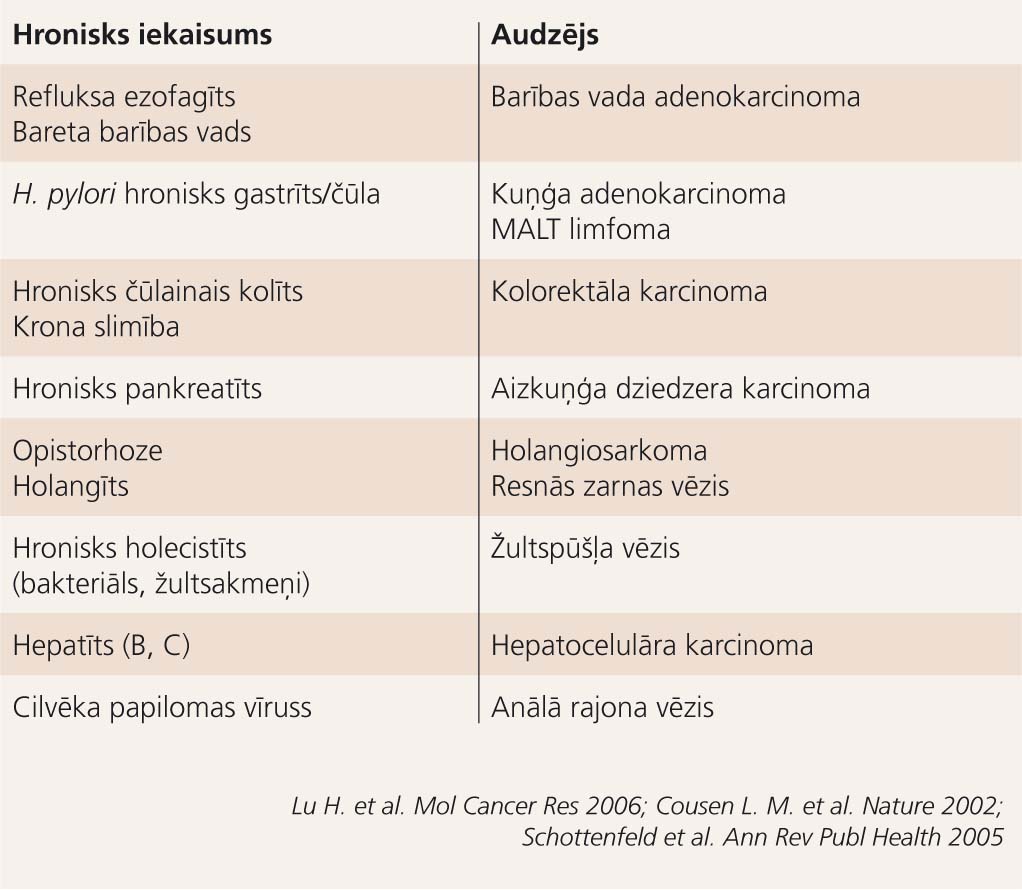
2. tabula
Ar hronisku iekaisumu saistīto gremošanas sistēmas orgānu ļaundabīgi audzēji
Gastroezofageāls atvilnis un barības vada vēzis
Pēdējo divdesmit gadu laikā pieaug barības vada adenokarcinomu skaits, kaut gan senāki epidemioloģiskie pētījumi liecina, ka gandrīz 90% no barības vada ļaundabīgajiem audzējiem bija plakanšūnu vēži, kas lokalizējās barības vada vidējā trešdaļā. Turpretī adenokarcinomas parasti lokalizējas barības vada apakšējā trešdaļā un cieši saistās ar Bareta (Barrett) intestinālo metaplāziju. [15] Virkne riska faktoru - tabaka, pastāvīgs atvilnis, barības vada atveres trūce, aptaukošanās, ģimenes anamnēze, antiholīnerģiskie līdzekļi, augsts pH - ir tie, kas veicina Bareta metaplāziju. [11]
Indivīdiem ar Bareta barības vadu un displāziju tajā adenokarcinomu barības vadā sastop 30-50 reizes biežāk nekā pārējā populācijā. Īpaši augsts risks ir tad, ja gļotādas izmaiņu garums pārsniedz 3 cm. [16; 17] Iepriekš minētie riska faktori saistās ar gastroezofageālu atvilni, kas savukārt izraisa hronisku ezofagītu; tad iespējama gļotādas čūlu veidošanās, barības vada metaplāzija, displāzija un visbeidzot - ļaundabīgs audzējs. Uz hroniska iekaisuma fona attīstījušās gļotādas transformācijas laikā veidojas arī virkne ģenētisku pārmaiņu: heterozigotijas zudums 9p un 17p hromosomā, aneuploīdija un poliploīdija. [18]
Helicobacter pylori un kuņģa vēzis
H. pylori ir gram- negatīva baktērija, kas dzīvo kuņģa corpus un antrum epitēlijā un ir viens no biežākajiem multifokālu atrofisku gastrītu, kuņģa un divpadsmit pirkstu zarnas čūlu, kā arī kuņģa adenokarcinomas cēloņiem. [19; 20] H. pylori infekcija vidēji divas reizes paaugstina risku saslimt ar kuņģa vēzi. Baktērija izraisa iekaisumu kuņģa gļotādā, procesā iesaistās mononukleārās šūnas, proinflamatorie citokīni, aktīvie skābekļa savienojumi, un epitēlijšūnās rodas DNS bojājumi. Kad izveidojies multifokāls atrofisks gastrīts, tālākā virzība ir viegli prognozējama: intestināla metaplāzija, displāzija, neoplastiska transformācija, invazīvs vēzis.
Vēl viens H. pylori onkogēno darbību ilustrējošs piemērs ir B šūnu limfomas (MALT limfomas) veidošanās ar gļotādu asociētajos limfoīdajos audos kuņģī. Kuņģī atrod apmēram 70% no šiem audzējiem. Zemas diferenciācijas MALT limfomas veidojas, atipiski mijiedarbojoties bakteriāliem antigēniem, saimnieka organisma autoantigēniem un saimnieka T šūnām. Šīs mijiedarbības rezultāts ir poliklonāla B limfocītu ekspansija, limfoīdo audu intensīva proliferācija un kavēta apoptoze. Molekulārā patoģenēze saistās ar imūnglobulīnu gēnu translokāciju un somatiskām mutācijām gēnos, kas saistītas ar B šūnu attīstību. [21; 22]
Hroniskas iekaisīgas resno zarnu slimības un kolorektāls vēzis
Hroniskas iekaisīgas zarnu slimības, čūlainais kolīts un Krona slimība saistās ar paaugstinātu resnās un taisnās zarnas vēža attīstības risku. Šo iekaisīgo slimību gaitā CD4+ T šūnu inducētā darbība iekaisuma atbildes reakcijas laikā netiek kontrolēta, izraisot pastiprinātu citotoksicitāti un imūno reakciju deviāciju, kas veicina epitēlijšūnu transformāciju. [23; 24]
Risks saslimt ar kolorektālu vēzi pieaug līdz ar iekaisīgās slimības ilgumu. 10-15 gadus pēc saslimšanas vēža risks pieaug par 0,5-1% gadā. Bieži šādiem pacientiem tiek konstatēti multicentriski audzēji. Nozīme ir arī patoloģiskā iekaisuma procesā iesaistītā zarnas segmenta garumam. Resnās un taisnās zarnas audzējus, ko saista ar iekaisīgām zarnu slimībām, biežāk diagnosticē gados jauniem pacientiem. Molekulārās izmaiņas zarnas gļotādā atrod jau pirms histoloģiskajām, un kolorektālā vēža molekulārajā patoģenēzē raksturīga šūnu ģenētiskā nestabilitāte, aneuploīdija, p53 mutācijas, DNS labotājgēnu promotoru metilēšanās u. c. [25; 26]
Vīrusu hepatīts B, vīrusu hepatīts C un hepatocelulāra karcinoma
Hroniska HBV un HCV infekcija saistās ar vairāk nekā 80% hepatocelulāro karcinomu (HCC) gadījumu pasaulē.
HBV ir DNS vīruss; kumulatīvais risks dzīves laikā saslimt ar hepatocelulāru karcinomu (HCC) hroniski inficētajiem tiek lēsts 10 līdz 20%. Latentais periods no inficēšanās brīža līdz HCC diagnozei variē no 20 līdz 50 gadiem. [27] Vīrusu inficētie hepatocīti mijiedarbojas ar T citotoksiskajiem limfocītiem un to sintezētajiem citokīniem, modulējot persistējošo iekaisuma procesu, šūnu apoptozi, nekrozi un reģenerāciju. Vīruss integrējas saimnieka genomā, arī protoonkogēnos. Iekaisuma procesā atbrīvojušies skābekļa savienojumi veicina DNS bojājumus šūnās un vīrusu integrāciju tajās. Vīrusu persistence vājina arī organisma pretaudzēja imūno atbildi. Jo ilgāku laiku vīruss persistē hepatocītos, jo lielāks risks saslimt ar HCC. [28; 29]
HCV ir RNS vīruss, vīrusa persistences mehānismi saistās ar tā mutācijas spējām un izvairīšanos no imūnās kontroles. 20-30 gadus pēc inficēšanās 2-4% šīs hroniskās aknu slimības pacientu attītās HCC. HCV infekcijas gadījumā audzēja attīstību biežāk novēro vīriešiem un cilvēkiem, kuri inficējas mūža otrajā pusē, kā arī tiem, kuri papildus inficējušies ar HBV vai HIV. Biežāk ar audzēju HCV gadījumā saslimst pacienti ar imūnsupresiju un alkoholiķi. [30; 31] Hepatīta vīrusa C inficētās aknās hronisks iekaisums saistās ar aktīvo skābekļa savienojumu izdali, DNS bojājumu, vīrusu integrāciju saimnieka šūnu genomā, pastiprinātu inficēto hepatocītu proliferāciju un to ģenētisko nestabilitāti. [32]
3. attēls
Iekaisuma veidi un to loma audzēja ģenēzē
Hronisks holecistīts un žultspūšļa vēzis
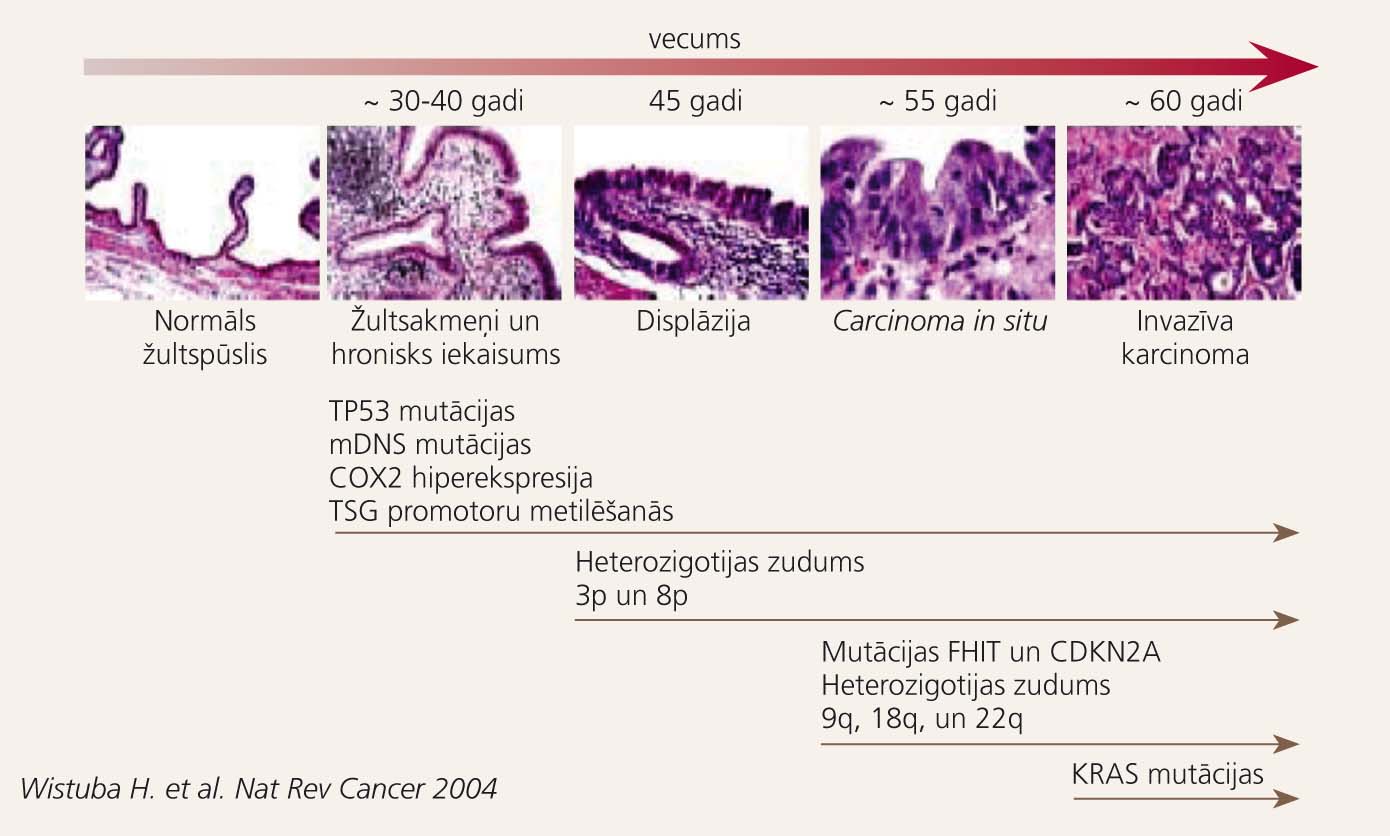
Viens no biežākajiem hroniska holecistīta iemesliem ir žultsakmeņi, kas veidojas holesterīna vielmaiņas traucējumu un žultspūšļa hipokinēzijas dēļ. Žults stāze lūmenā bojā gļotādu, veicinot intracelulāro enzīmu atbrīvošanos un iekaisuma mediatoru (prostaglandīnu, fosfolipāžu, citokīnu) kaskādes aktivēšanos. Tiek izjaukts šūnu proliferācijas un apoptozes līdzsvars, pieaug ļaundabīgā audzēja attīstības iespēja. Žultsakmeņu izraisīts hronisks holecistīts palielina risku saslimt ar žultspūšļa vēzi 15-25 gadus pēc holecistīta diagnosticēšanas. [33] Ģenētisko izmaiņu kaskāde, kas noris, morfoloģiski mainoties žultspūšļa epitēlijšūnām, redzama 4. attēlā.
4. attēls
Žultspūšļa ļaundabīga audzēja attīstības etapi
1863. gadā izteikto Virhova hipotēzi, ka ļaundabīgie audzēji veidojas iekaisuma vietā, kārtējo reizi 2006. gadā apstiprināja ASV zinātnieku grupa, publicējot pārskatu par pētījumu, kas saistās ar iekaisīgām slimībām žurnālā Cancer for Clinicians: „Slimības, kas pēc savas klīniskās un patoloģiskās ainas klasificējamas kā iekaisīgas, uzskatāmas par vēža prekursoru..."